-
CRISPR篩選和糖蛋白質組學聯合研究發現新型WNT通路的N-糖基化調控機制
發布時間: 2025-11-13 點擊次數: 390次——跟著Science學習如何進行N-糖基化修飾的機制研究

美國斯坦福大學Rajat Rohatgi研究組發現,受調控的N-糖基化控制伴侶蛋白功能和受體轉運。這與將N-糖基化視為常規維護功能的普遍看法相反。研究人員通過N-糖肽組學分析揭示了內質網中一個調控N-糖基化的新途徑:該途徑由OST-A與HSP90B1(一種膜受體的ER伴侶蛋白)和CCDC134(一種ER腔內蛋白)組成,在HSP90B1轉運到ER的過程中,其N端肽段引導了包含CCDC134和OST-A的易位復合體的組裝,該復合體在折疊期間保護HSP90B1,防止其過度糖基化和降解。該通路對于WNT信號傳導至關重要,其功能障礙可能導致骨發育障礙。這一研究結果于2024年11月8日發表在Science雜志上,題為“Regulated N-glycosylation controls chaperone function and receptor trafficking"。
研究結果
基于CRISPR篩選激活WNT/β-catenin信號的基因
使用基于熒光的轉錄報告基因,在人類細胞系(RKO)中進行了全基因組的功能缺失CRISPR/Cas9篩選,以鑒定響應WNT3A激活WNT/β-catenin信號所需的基因。篩選發現了許多已知的WNT信號成分: β-catenin, LRP6 (WNT配體的共受體),以及兩種ER伴侶,MESD和HSP90B1,促進LRP5和LRP6 (LRP5/6)在ER中的折疊。篩選還發現了三個與WNT信號不明確相關的基因:CCDC134、STT3A和OSTC。
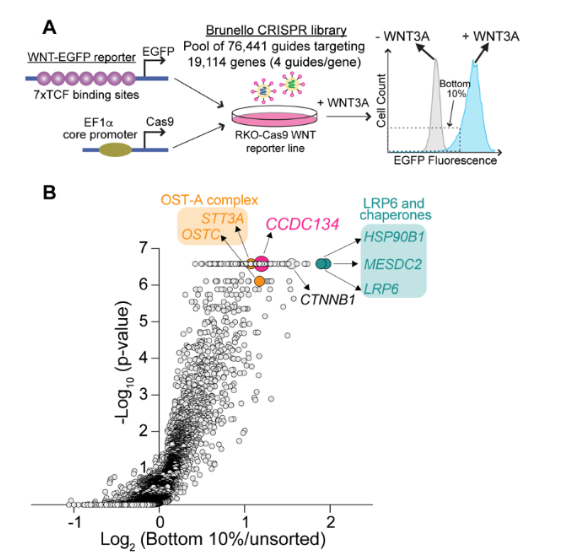
圖1. CRISPR/Cas9篩選鑒定鑒定響應WNT3A激活WNT信號傳導的陽性調節因子。
內質網伴侶HSP90B1的高度糖基化調控
敲除STT3A(而不是STT3B)導致HSP90B1的高度糖基化和去穩定化。CCDC134和OSTC的敲除也導致HSP90B1的高度糖基化和不穩定,此外,STT3A、CCDC134或OSTC的缺失降低了細胞表面WNT共受體LRP6的豐度。高度糖基化的HSP90B1不能折疊成功能蛋白,導致其被ERAD機制標記和降解。N-糖蛋白學顯示STT3A和CCDC134與HSP90B1的N-糖基化具有高度特異性和一致性。

圖2. 內質網蛋白網絡對HSP90B1的N-糖基化和WNT信號的調控。
內質網中CCDC134對細胞表面WNT信號的調控
多種小鼠和人細胞系中CCDC134的敲除導致HSP90B1高度糖基化和不穩定。回補實驗表明,使用多xi環素誘導系統可以恢復CCDC134的表達,抑制HSP90B1高度糖基化并恢復其豐度。此外,CCDC134的共表達抑制了其過表達引起的HSP90B1高度糖基化,并且在ER應激下對HSP90B1具有劑量依賴性的保護作用。CCDC134-/-細胞表面LRP5/6豐度明顯降低。CCDC134-/-細胞對WNT配體的反應遠低于野生型細胞。CCDC134通過控制LRP5/6 (WNT配體的專性共受體)到細胞表面的運輸來調節內質網中的WNT信號。CCDC134-/-患者的原代成纖維細胞表現出HSP90B1高度糖基化,細胞表面LRP6豐度降低,WNT信號通路受損,所有這些缺陷在CCDC134重新表達后被逆轉。

圖3. 在攜帶CCDC134突變的人類患者中 HSP90B1高度糖基化和LRP6。
CCDC134、OST-A和HSP90B1組成的信號通路
與CCDC134或HSP90B1的缺失類似,敲除STT3A(但不包括STT3B)也顯著降低了細胞表面LRP6和WNT信號的強度。值得注意的是,HSP90B15N的表達足以wan全挽救STT3A-/-細胞中的WNT信號,盡管當OST-A功能喪失時,數百種膜和分泌蛋白的N-糖基化會發生改變。與CCDC134-/-細胞相比,STT3A-/-細胞中HSP90B1 N-糖基化有顯著差異。前者所有HSP90B1被高度糖基化,而后者只有一小部分被高度糖基化。即使CCDC134豐度大量增加,也無法抑制STT3A-/-細胞中的HSP90B1高度糖基化。這些數據表明,OST- B(STT3A-/-細胞中唯yi的OST復合物)在所有序列中默認wan全N-糖基化HSP90B1,并且不能被CCDC134調節。在STT3B-/-細胞中,CCDC134的缺失導致HSP90B1高度糖基化,這表明CCDC134可以調節OST-A。在缺乏CCDC134的情況下,與OST-B相比,OST-A對HSP90B1的N-糖基化效率較低。
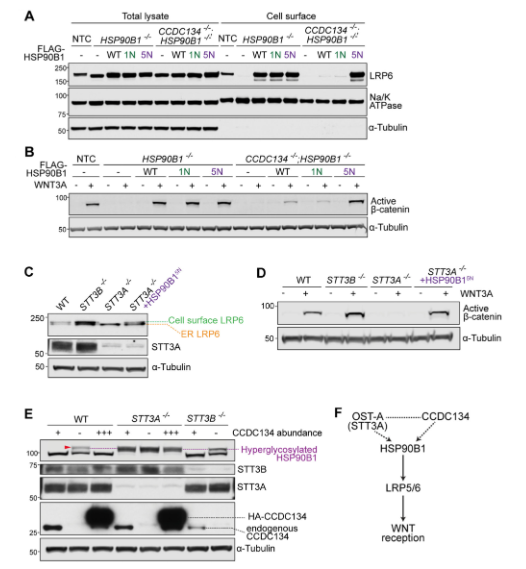
圖4. 高度糖基化的HSP90B1調控WNT信號通路
HSP90B1中的N端非結構化肽調節其自身的N-糖基化修飾
系統缺失分析表明,HSP90B1的1-93氨基酸,包括ER信號序列和所有前N段,可以影響M結構域遠端序列的N-糖基化修飾。與STT3A-/-細胞中的HSP90B1相似,HSP90B1- T44A突變體也對CCDC134調控具有抗性。 HSP90B1的N端1~93個氨基酸,預計在很大程度上是非結構化的,具有兩個自主的,可轉移的特性:它損害在同一多肽中緊隨其后的序列的N糖基化,也賦予CCDC134敏感性。HSP90B1的高度糖基化是由其自身N端、CCDC134和OST-A之間的復合物調節的模型,該復合物在其翻譯和轉運到內質網時組裝。
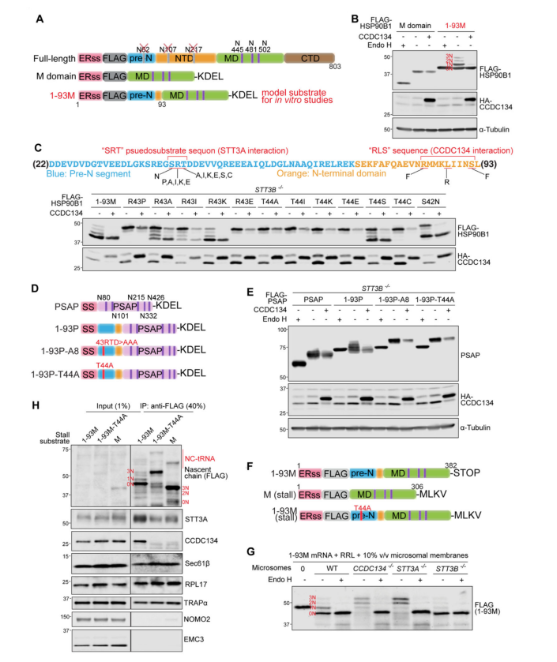
圖5. HSP90B1的前體N段通過招募CCDC134到含有OST-A的易位子來抑制其自身的N-糖基化。
OST-A作用機制
OST-A的糖基轉移酶活性對于抑制HSP90B1過度糖基化并非必需。這一發現挑戰了我們對OST-A功能的傳統理解,提示OST-A可能通過非酶活性的方式參與蛋白質的折疊和質量控制。研究人員通過構建OST-A的突變體,并在細胞中表達這些突變體,發現即使在OST-A的糖基轉移酶活性被抑制的情況下,HSP90B1的糖基化仍然受到抑制,表明OST-A可能通過其他機制調控HSP90B1的糖基化。OST-A可能不是作為一種酶起作用,而是作為一種支架起作用。

圖6. HSP90B1的穩定性和WNT信號傳導不依賴OST-A的寡糖轉移酶活性。
文章總結
一、主要方法
· 利用細胞生物學方法(如免疫印跡、共沉淀、顯微鏡定位)分析蛋白修飾與相互作用。
· 通過突變體(改變N-位點或糖基化酶活性)、化學抑制劑(如抑制糖鏈修剪或加成)調控糖基化過程。
· 功能性測定包括受體在細胞表面的表達量、配體結合/信號轉導能力及降解速率等。
· 質譜分析糖基化位點、以及生化方法分析糖鏈結構變化。
二、核心發現
1. N-糖基化是可調控的,并且其狀態(是否附有完整糖鏈或是否已被糖苷酶/糖轉移酶修飾)決定了伴侶蛋白對底物的識別與結合強度。
2. 伴侶蛋白(如calnexin/calreticulin)對特定糖基化狀態的偏好影響其與新生受體蛋白的相互作用時間,從而改變折疊效率與質量控制決策(繼續折疊/進入降解通路)。
3. 改變N-糖基化(通過基因突變或化學干預)會直接影響某些受體(可能為膜受體或分泌受體)的成熟與從ER到高爾基體及細胞膜的轉運,導致細胞表面受體量和功能發生變化。
4. 該調控機制在細胞對壓力或生理信號響應時可能被動員,以調整細胞表面受體數量或調節分泌蛋白的輸出,從而參與更廣泛的細胞功能與適應性反應。
三、結論與意義
· N-連接糖基化不僅是結構性修飾,也是動態調控蛋白命運的關鍵信號。
· 通過控制伴侶蛋白與底物的相互作用,糖基化狀態決定受體是否通過質量控制并正確定位到細胞表面。
· 理解這個調控軸有助于解釋某些遺傳病、代謝或分泌失調的分子機制,并為開發針對糖基化或伴侶系統的小分子干預提供理論依據。
四、后續研究方向
· 識別更廣泛的受體/底物譜及其敏感的糖基化位點。
· 解析不同細胞類型或生理狀態下糖基化調控的機制(例如糖酶、轉移酶的調控)。
· 開發藥物以修復因糖基化異常導致的蛋白折疊或定位缺陷。



